Molekülorbital-Datenbank
Diese Datenbank verfügt über ein Web-basiertes Interface und beinhaltet eine stetig wachsende Anzahl an organischen π-konjugierten Molekülen mit detaillierten Informationen über ihre Molekülorbitale. Sie wurde mithilfe des Python Pakets "Atomic Simulation Environment" (ASE) erstellt und nutzt den Dichtefunktionaltheorie-Code NWChem zur Berechnung. Die Molekülorbitale in dieser Datenbank können außerdem direkt über das Pythonprogramm "kMap.py" für Simulationen in der Photoemissionsorbitaltomographie verwendet werden. Wenn Sie Vorschläge für bestimmte Moleküle haben oder selbst Beiträge zur Erweiterung der Datenbank liefern wollen, kontaktieren Sie mich bitte über E-Mail.
kMap.py
@Code, @Wikipedia, @YouTube, (Built Distribution für Windows 10 verfügbar auf Anfrage)
kMap.py ist ein in Python-Programm zur Simulation und Datenanalyse in der Photoemissionstomographie. Es bietet eine einfach zu bedienende grafische Benutzeroberfläche (basierend auf PyQt5), um Photoemissions-Impulskarten von Molekülen zu simulieren und erlaubt einen direkten Vergleich zwischen der Simulation und dem Experiment. Zu diesem Zweck stellt kMap.py verschiedene Werkzeuge zur Verfügung, wie linien- oder regionsbeschränkte Intensitätsprofile/diagramme oder Interpolationsmethoden. Simulationsparameter (wie Orientierung des Moleküls, kinetische Energie des Endzustands oder Polarisierung des eintreffenden Lichts) können angepasst werden und ein Interface zur Least-Square Fit-Bibliothek "LMfit" erlaubt die Bestimmung der optimalen Parameter durch Vergleich von Simulation und Experiment.
Publikation: Brandstetter et al., kMap.py: A Python program for simulation and data analysis in photoemission tomography, Computer Physics Communications 263, 107905 (2021)
ARUPS@VASP
Der ARUPS@VASP Code ist ein Fortran-Code zur Simulierung der winkelaufgelösten Photoemissionsintensität (ARUPS), basierend auf dem "one-step"-Modell der Photoemission. Als Anfangszustand werden Kohn-Sham Orbitale einer VASP-Rechnung (WAVECAR File) verwendet. Als Endzustand kann entweder eine ebene Welle oder eine gedämpfte ebene Welle verwendet werden. Das Programm simuliert ARUPS Intensitätsverteilungen entweder als "band-maps" I = I(k, Eb) für eine gewisse Emissionsebene, als Impulskarten bei konstanter Bindungsenergie, "k-map" I = I(kx, ky), oder als sogenannter "constant initial state" (CIS) scan I = I(k, E_photon) als Funktion der Photonenergie. Mehr Informationen über die Methode sowie Ergebnisse ihrer Anwendung finden Sie hier:
Publikation: D. Lüftner et al., Understanding the photoemission distribution of strongly interacting two-dimensional overlayers, Phys. Rev. B 96, 125402 (2017).

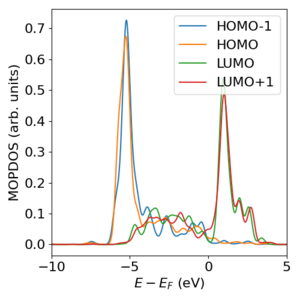
MOPDOS@VASP
Der MOPDOS@VASP code ist ein Fortran-code, der die sogenannte "molecular orbital projected" (MOP) density of states (DOS) berechnet, also die elektronische Zustandsdichte eines Systems projiziert auf die Orbitale eines Teilsystems. Konkret kann damit z.B. bei der Berechnung der Grenzfläche eines Moleküls mit einer Oberfläche die Zustandsdichte der Grenzfläche auf die Molekülorbitale des isolierten Moleküls projiziert werden. Der code benötigt die Wellenfunktionen der beiden Systeme (Gesamtsystem und Teilsystem) in Form der WAVECAR-files einer VASP-Rechnung.
Publikation: D. Lüftner et al., Understanding the photoemission distribution of strongly interacting two-dimensional overlayers, Phys. Rev. B 96, 125402 (2017).